


From SMILES to Graphs: The Next Frontier in ML-Driven Cheminformatics
Since the early days of computing, scientists and engineers have been inventing ways to represent molecules in a language that machines can understand. Whether it’s for searching chemical databases, running simulations, or applying the latest machine learning (ML) methods, these digital representations form the backbone of modern computational chemistry.
Early on, molecular file formats served a simple purpose: store chemical structures so that researchers could exchange and analyze data without losing important details. Over the past 30 years, numerous formats and conventions have emerged, each aiming to tackle unique
A Quick Look at SMILES
Among the most popular ways to describe a molecule in text form is SMILES (Simplified Molecular Input Line Entry System). SMILES encodes the structure of a molecule as a string, typically generated by performing a depth-first traversal of the molecular graph.
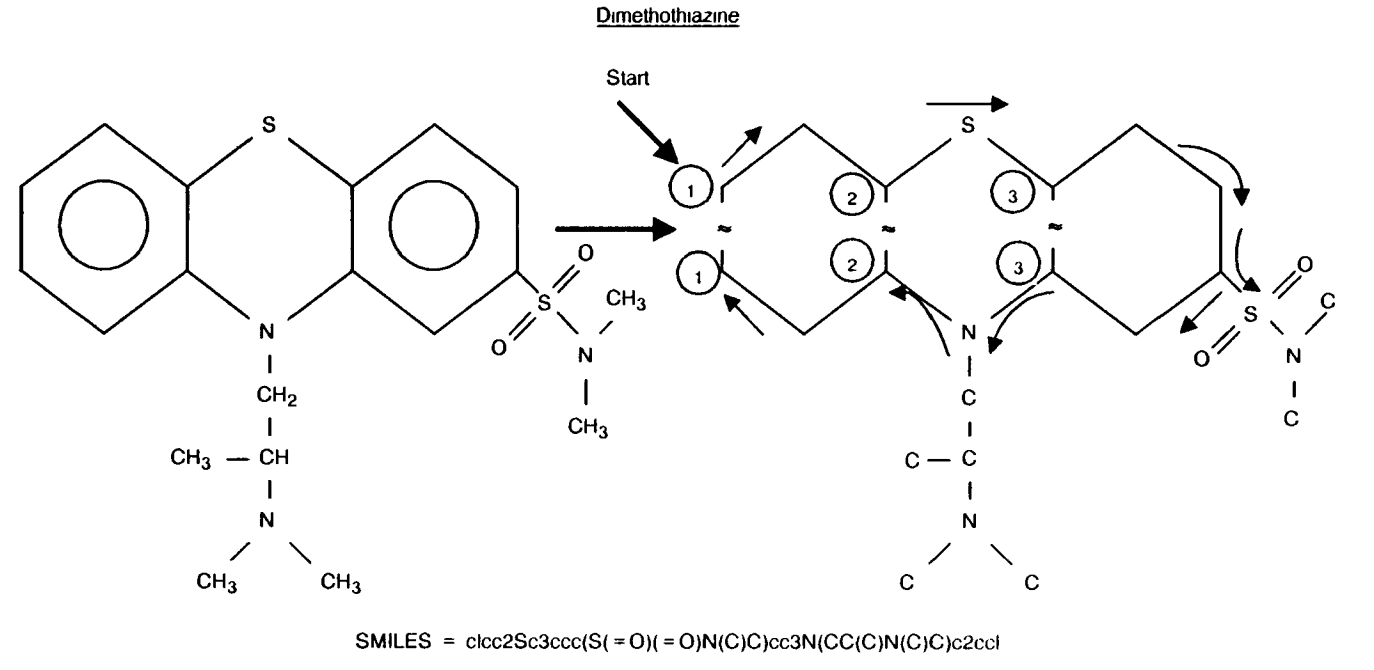
Image Source: Anderson E, Veith GD, Weininger D (1987). SMILES: A line notation and computerized interpreter for chemical structures (PDF). Duluth, MN: U.S. EPA, Environmental Research Laboratory-Duluth. Report No. EPA/600/
Why SMILES became popular:
Simplicity: It’s concise, human-readable, and easy to copy-paste between software.
NLP Synergy: Its string-based format tempted researchers to apply Natural Language Processing (NLP) techniques (like Transformers) developed for text to molecular problems.
Despite these advantages, SMILES has some well-known drawbacks:
Multiple Dialects: Different software may produce different SMILES for the same molecule (though canonicalization tools exist).
Lack of Rich 3D Detail: SMILES focuses on connectivity; capturing rotamers, subtle chirality, and conformers is limited or requires extra layers of complexity.
Sensitivity to Atom Order: The SMILES string you get depends on the chosen starting atom. While you can canonicalize, this step is itself non-trivial and sometimes unreliable for large or unusual molecules.
As a result, when it comes to complex
Graph-Based Representations
Graph-based formats describe a molecule in terms of nodes (atoms) and edges (bonds). In the most basic version, each atom is a node tagged with attributes like element type or charge while edges store bond orders (single, double, triple and others) and other relevant information. More advanced versions can also include 3D coordinates for each atom.
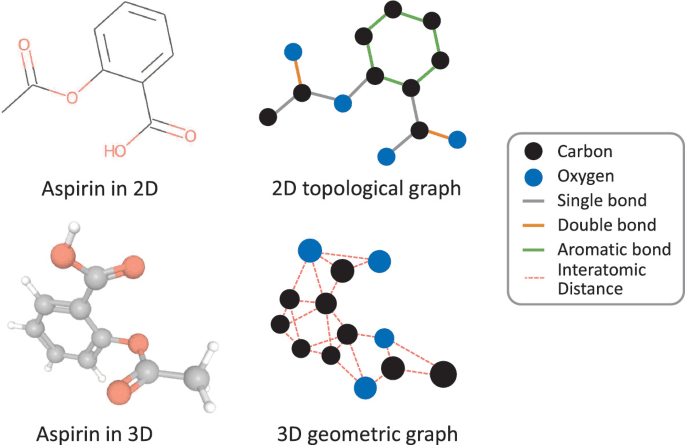
Image Source: Wang, Y., Li, Z., Barati Farimani, A. (2023). Graph Neural Networks for Molecules. In: Qu, C., Liu, H. (eds) Machine Learning in Molecular Sciences. Challenges and Advances in Computational Chemistry and Physics, vol 36. Springer, Cham. https://doi.org/10.1007/978-3-031-37196-7_2
Why graphs shine in cheminformatics:
Flexibility: They naturally encode connectivity, stereochemistry, and can even incorporate 3D coordinates for each node.
Rich Data Structure: Graphs are better at handling ring systems, branching, and complex topologies without losing crucial information.
GNNs: Graph Neural Networks (GNNs) are designed to process graph-structured data, making them a more direct match for molecular tasks.
Beyond 2D: The importance of 3D
Many chemical
- Predict detailed 3D conformations, which is vital for tasks such as structure-based drug design.
- Capture stereochemical nuances and subtle conformational preferences.
- Generate brand-new 3D structures that meet specific property constraints.
When SMILES isn’t enough
In some scenarios, simple connectivity just doesn’t cut it. This is especially true in generative chemistry, where you might want to:
- Design molecules that fit a particular 3D shape or binding pocket.
- Optimize properties like solubility, toxicity, or reactivity, which can be strongly influenced by 3D conformation.
- Ensure correct stereochemistry in drug-like molecules, where the wrong chirality can make or break a compound’s efficacy.
Using SMILES in generative tasks can lead to problems like invalid or duplicated molecular structures due to the complexities of string-based generation. Graph-based models handle these challenges more gracefully because they operate directly on the underlying molecular
A Glimpse into the Future: EDM and Beyond
One of the most exciting frontiers in graph-based molecular modeling is the use of diffusion models, sometimes referred to under acronyms like EDM (Equivariant Diffusion Models). These models start with random “noise” in 3D space and iteratively learn to “denoise” until a coherent molecular structure emerges
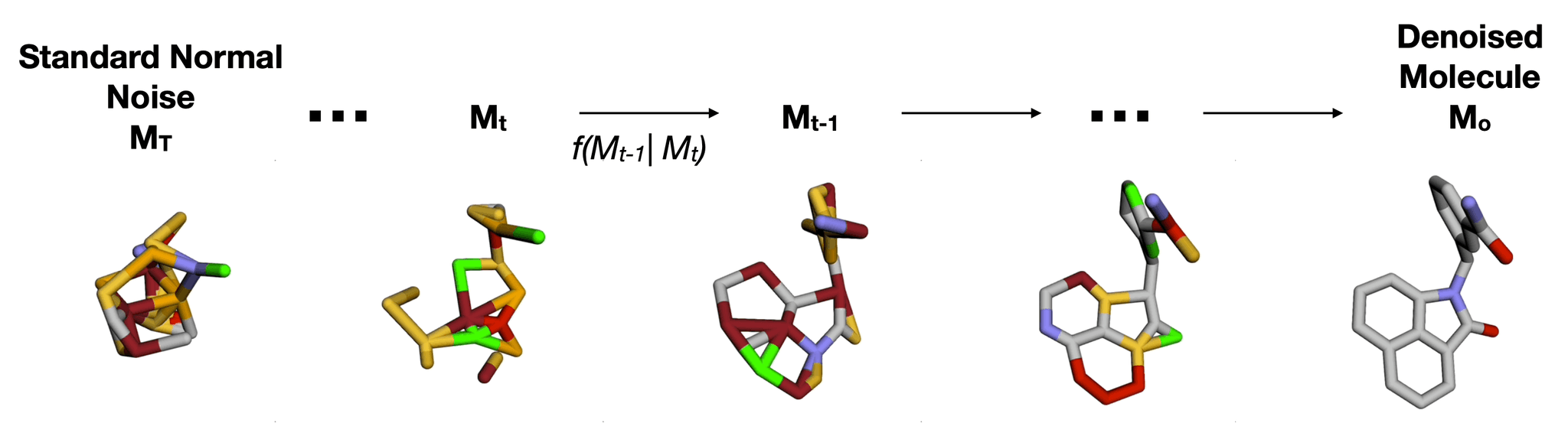
*f(
Partnered with Nebius, Quantori has taken on a project to develop an EDM framework to generate molecules similar to a given shape. By leveraging shape descriptors for contextual generation and training of the equivariant graph diffusion network, the model learns to generate molecules that fit the same 3D spaces as the target structures. The atom coordinates generated by the EDM model are then passed to a pre-trained Structure Seer model, which predicts where and which bonds are located.
This approach allows researchers to rapidly ideate on potential candidate
Our EDM model was trained on a set of small molecules from the ChEMBL database, using a cluster on the Nebius cloud equipped with 8 H200 Nvidia GPUs. The quick provisioning and easy configuration of these resources enabled us to set up training and evaluation with minimal effort, while the GPU capacity allowed training on a dataset containing 1.6 million compounds, ultimately leading to better model quality. During training, we generated valid random conformers for each molecule, supplied them to the model, and guided it toward geometrically similar outputs using shape-descriptors.
Imagine a molecule being formed from a cloud of seemingly random points. Step by step, these points move into place, bonding with each other to produce a real, chemically sound 3D structure. The final result: a new molecule ready to be tested for a given property or shape constraint.
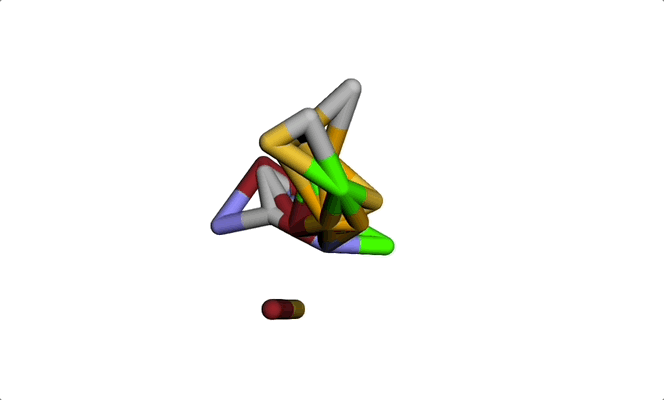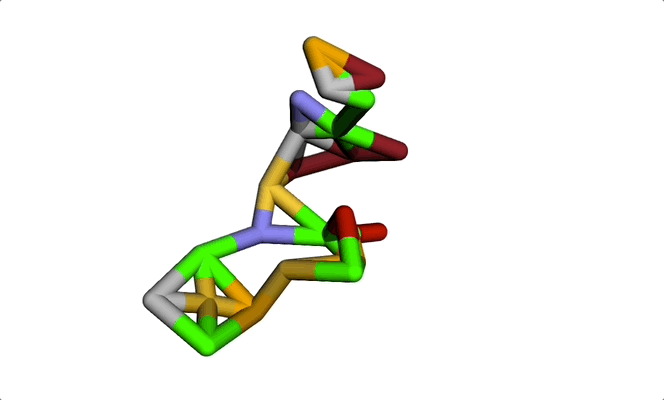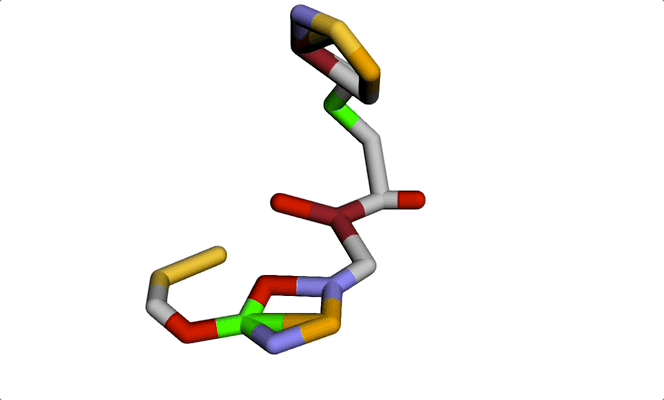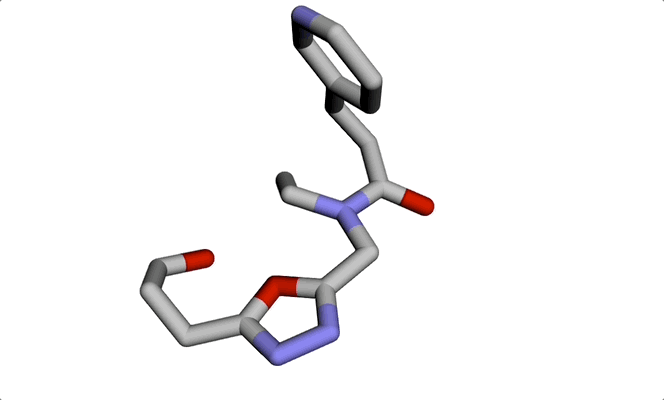
This visually striking process showcases how diffusion models and graph-based representations bring molecule generation to
After 1,500 epochs (about 13.5 days) of training, the results proved promising, showing that the selected shape descriptors captured the necessary information to drive valid 3D conformer generation with a shape similar to a reference. The generation of 50 samples on the GPU without paralellisation or additional optimisation takes around
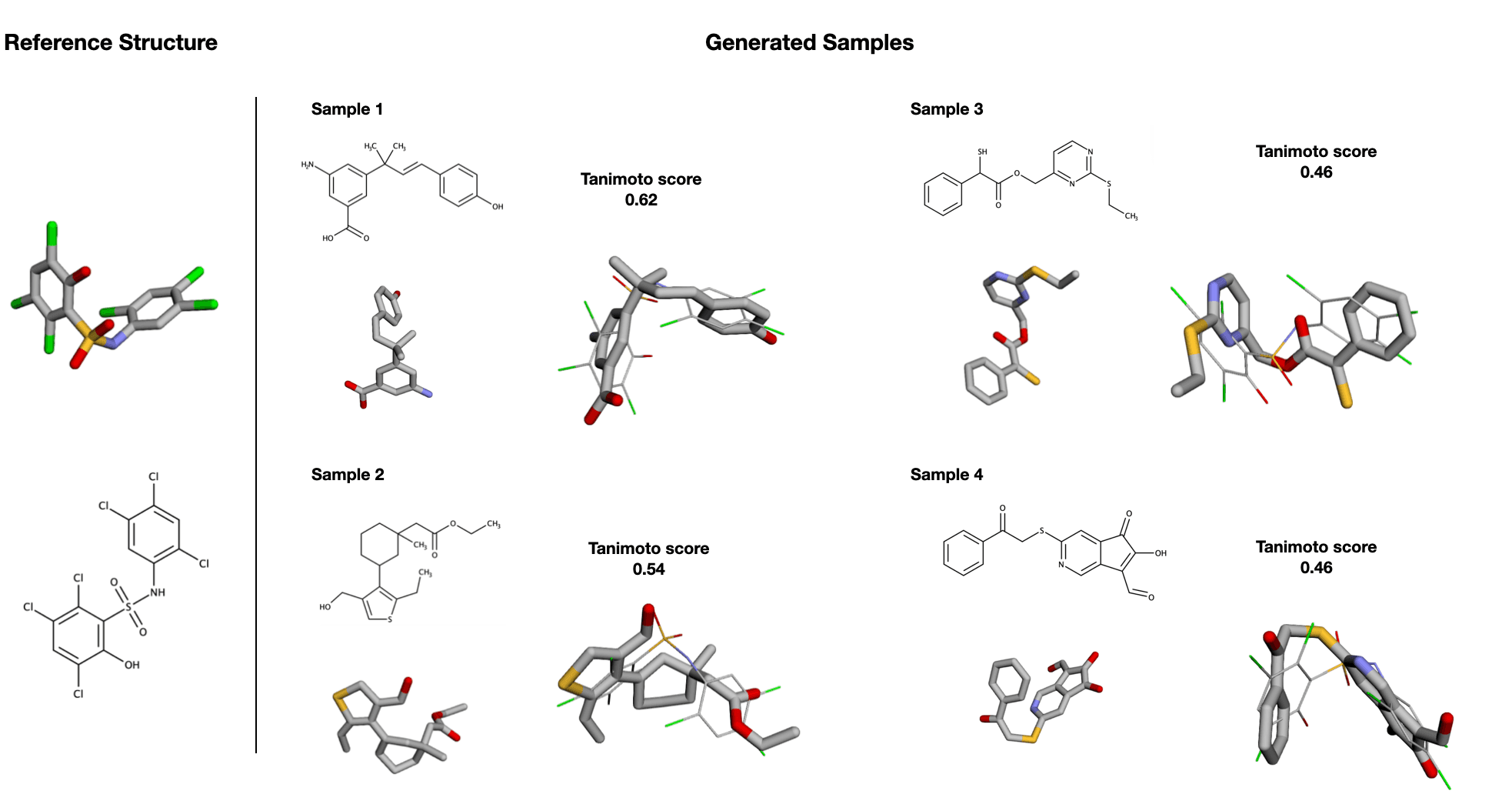
Conclusion
While SMILES revolutionized molecular representation by offering a simple, linear encoding well-suited for certain cheminformatics tasks, it falls short for many cutting-edge
From molecular property prediction to in silico drug design, the future of